Detecting Natural Selection (Part 4)
Phylogenetics and Relative Rates
This is the fifth of multiple postings I plan to write about detecting natural selection using molecular data (ie, DNA sequences). The first post contained a brief introduction and can be found here. The second post described the organization of the genome, and the third described the organization of genes. The fourth post described codon based models for detecting selection.
The simple codon based model for detecting natural selection that I described previously (dN/dS) involves comparing two homologous sequences. If we have three or more sequences, we can create a rooted phylogeny, and four or more sequences allow us to create an unrooted phylogeny. With the analysis of dN and dS we were not concerned with which lineage the substitutions occurred on. In our relative rate analysis we will be determining were in the tree (on which branch) the substitutions occurred.
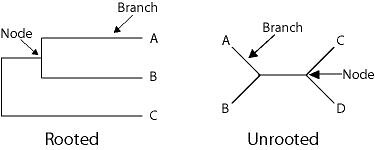
I will not get into the detail regarding different algorithms for creating phylogenies, and I will assume that we already know the evolutionary relationship of the sequences we are comparing. If you are interested in learning more about how phylogenies are created, I would recommend starting with this book and following the literature citations therein. I will point out that the length of the branches represents the number of substitutions that have accumulated along a particular lineage. Phylogenies can be created with either DNA sequences or translated protein coding sequences (amino acid sequences) depending on if the sequences are closely related or not. (DNA sequences are preferred for closely related sequences because they evolve faster and accumulate more substitutions in a shorter period of time, while amino acid sequences are preferred for more distantly related sequence because they evolve slower.)
Recall from our codon based comparisons that we have, essentially, three different selective scenarios:
- Neutral evolution - a sequence is evolving without the constraint or influence of natural selection
- Purifying selection or selective constraint - natural selection is acting as a conservative force, restricting the evolution of a sequence
- Positive selection - natural selection is driving the evolution of a sequence, causing it to evolve faster than the neutral expectation
Relative rate tests compare the selective constraint along two lineages of a phylogeny. Assuming all other factors are equal (ie, population size, mutation rate, gametogenesis, generation time), if the selective constraint along both lineages is equal, the branch going from sequence A should be of equal length to the branch leading to sequence B. If, however, the selective constraints differ, the branch lengths should be unequal.

If there are more substitutions along one lineage than the other, we must invoke some explanation for these differences. In some cases, differences in rates can be attributed to life history differences of the species from which the two sequences come. For example, mice have much shorter life spans than humans, and this has been used to explain differences in rates of evolution between the two lineages. Body size and metabolic rate can also affect rates of evolution, as can whether the sequence is from an X-chromosome or autosome -- autosomes are found equally in males and females, whereas X-chromosomes are disproportionately found in females, and male gametogenesis involves more cell divisions (more potential for mutation) than female gametogenesis.
We can control for the effects of life history on rate differences by selecting two genes from a single genome (ie, duplicate genes) or obtaining sequences from organisms with similar life histories. If we are interested in comparing sequences from two species with known life history differences, we can sample multiple sequences from each of those species (as well as our third species). Life history should affect all sequences equally, whereas selection should only affect a subset of the sequences.
If we observe differences in rates along two lineages after controlling for other variables, we conclude that the selective constraints along the two lineages differ. The difference can be due to increased purifying selection along the slowly evolving lineage (the shorter branch) or positive selection along the rapidly evolving lineage (the longer branch). Distinguishing between these two hypotheses requires more information (such as difference in synonymous and non-synonymous substitutions). Some of the other assays for natural selection that I will describe can also be used to discriminate between increased selective constraint and positive selection.
I am not sure if I will post another entry on comparative and phylogenetic analyses, or if I will move on to discussing nucleotide polymorphism in my next post. If you have any suggestions, or further questions, please post them in the comments.
posted by RPM @ 1:56 PM
0 comments
![]()
![]()











0 Comments:
Post a Comment
<< Home